Behind the Scenes of a Successful FDA 510(k): PathPresenter’s Clinical Viewer Journey

by Brian Matcheski
Regulatory Manager, PathPresenter
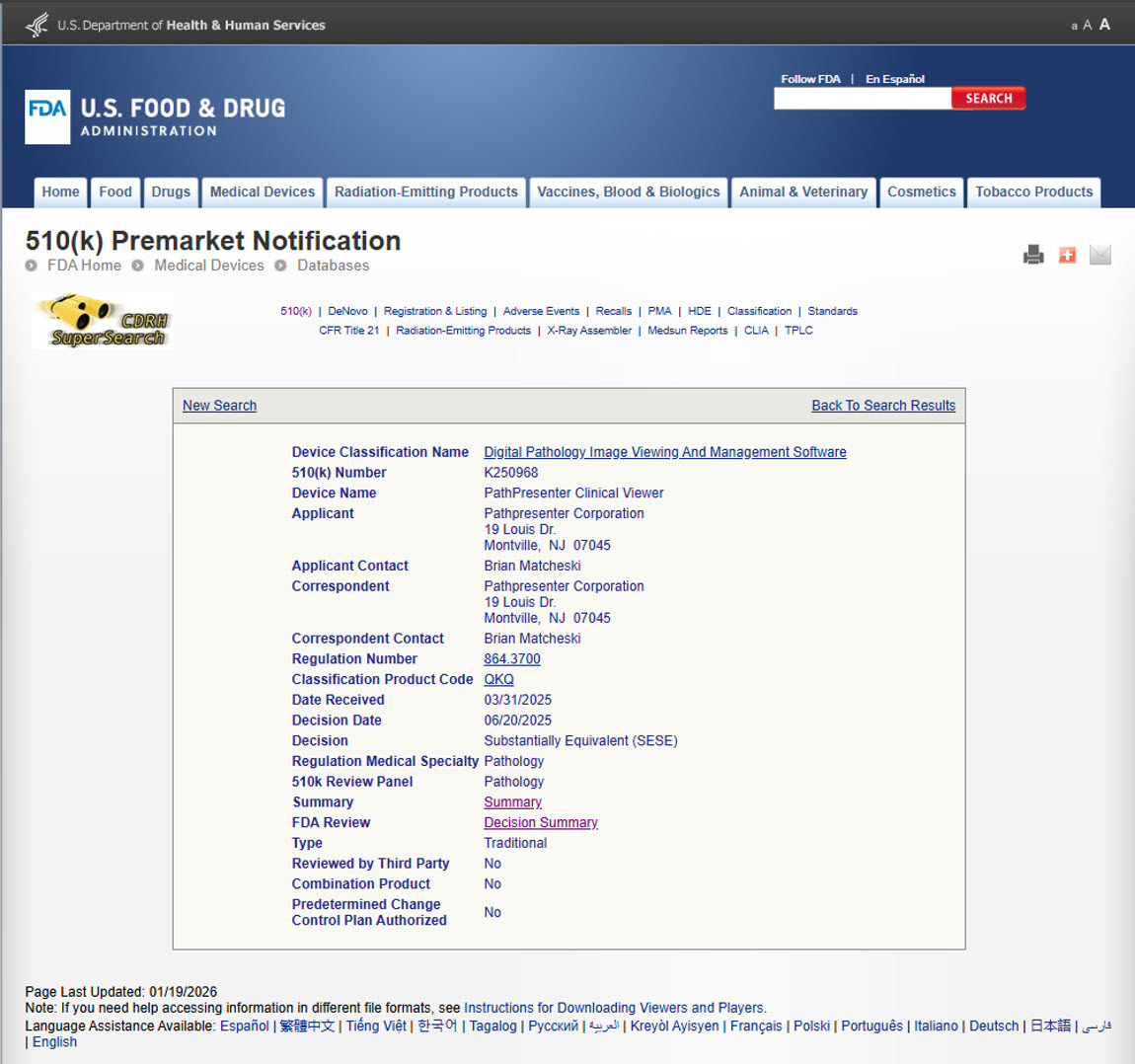
FDA 510(k) clearance is often described as a milestone in digital pathology. What’s discussed less is how you actually get there, especially for software-only image viewers, where regulatory expectations differ meaningfully from scanners and other hardware-based systems.
With the recent FDA 510(k) clearance of the PathPresenter Clinical Viewer, we wanted to share some perspective on our journey: what mattered most, what surprised us, and why a disciplined approach to the process ultimately pays dividends. This isn’t a detailed how-to guide, and there are no shortcut hacks to share (with the possible exception of “get good advice from a qualified regulatory expert with experience in digital pathology”). But for those involved in navigating FDA clearance for digital pathology software, these insights from our journey may prove interesting.
Why FDA Clearance Matters in Digital Pathology

As digital workflows in pathology move from research into routine clinical use, regulatory oversight becomes essential to building trust and enabling adoption. FDA regulation of digital pathology software ensures that clinical tools meet standards for safety, effectiveness, and reliability.
The FDA has several pathways for market clearances, but to date, digital pathology devices have concentrated on just two: de novo and 510(k). The de novo (Latin for “new”) pathway is used for new devices with no existing precedent (or predicate). The 510(k) pathway applies when a new device can demonstrate substantial equivalence to an existing, legally marketed predicate. Clearly, the 510(k) pathway is only available when a predicate already exists; it clears a new component to do an equivalent job in an existing system. Not surprisingly, de novo clearances typically involve greater regulatory uncertainty, broader risk justification, and longer timelines than 510(k).
Neither clearance pathway represents FDA “approval,” nor do they validate clinical outcomes, but clearance is a critical prerequisite for deploying software in regulated diagnostic environments.
For pathologists and labs, FDA clearance provides confidence that a viewer can be used in primary diagnostic workflows. For healthcare systems and enterprise customers, it reduces risk and enables broader clinical integration. For PathPresenter customers, our 510(k) clearance unlocks the ability to deploy our Clinical Viewer in regulated clinical settings with confidence.
Understanding Classification, Intended Use, and Predicates
One of the earliest and most important steps in the process for PathPresenter was clearly defining intended use. Digital pathology viewers are regulated differently from whole slide imaging (WSI) scanners. While scanners capture the image, viewers render, display, and enable interpretation of that image, and the FDA evaluates them separately.
That distinction drives selection of the appropriate predicate device. Substantial equivalence hinges not only on features, but on intended use, performance characteristics, and risk profile. Getting alignment here early set the foundation for the entire submission.
Planning to Succeed: Following the FDA’s Guidance
One of the best decisions we made was to anchor our submission directly to FDA guidance documents, including:
- Technical Performance Assessment of Digital Pathology Whole Slide Imaging (WSI) Devices
- Content of Premarket Submissions for Device Software Functions
- Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions
These documents do more than list requirements, they provide a window into how FDA reviewers think about risk, safety, and effectiveness. By structuring our submission around this guidance, we weren’t guessing what the FDA wanted to see. We were aligning with how the agency approaches review.
A strong 510(k) submission is not about volume. It’s about completeness, traceability, and clarity.
Working with FDA Reviewers
Going into the review, we were cautious. In mid-2025, widely reported austerity measures affecting HHS and the FDA raised concerns across the industry about review timelines and resource constraints.
In our experience, those concerns did not materialize. The FDA’s feedback throughout the review was timely, thoughtful, and constructive. Importantly, our submission was reviewed and cleared within the FDA’s stated 90-day service level.
That experience reinforced our confidence in both the process and the people behind it. Even in a challenging operating environment, the review team remained focused on scientific rigor, patient safety, and predictable execution.
Investing Early Where It Mattered Most: Pixel-Wise Image Comparison
For software-only digital pathology viewers, one of the most scrutinized aspects of a 510(k) submission is pixel-wise image comparison to the predicate viewer. This is an area where other OEMs have historically faced delays.
For a clinical viewer, the primary safety risk is not image acquisition but image presentation. Even small rendering differences, such as altered color balance, contrast, or interpolation, could theoretically influence a pathologist’s interpretation. Pixel-wise comparison evaluates whether the same whole slide image (WSI), when rendered by two different viewers, produces image outputs that are effectively the same at the pixel level. The goal is to show that the subject viewer does not introduce image artifacts, distortions, or rendering differences that could impact clinical interpretation.
We anticipated scrutiny in this area and invested significant effort upfront. That meant carefully defining our comparison methodology, validating our approach, and presenting results that were clear, reproducible, and defensible.
That investment paid off. When FDA reviewers conducted their own image evaluations, they were able to quickly confirm that our viewer met performance expectations, without prolonged back-and-forth. It was one of the most valuable early investments we made in the process.
Responding Quickly and Completely to FDA Feedback
Even with a thorough submission, FDA reviewers are trained to identify areas of potential risk or ambiguity (and they should). We treated every FDA comment as an opportunity to strengthen the submission.
We made our responses timely, direct, and supported by data. There are no shortcuts here. That responsiveness helped maintain momentum and avoid unnecessary review cycles, which can otherwise add weeks or months to the process.
Treating Pre-Submission Feedback as a Contract
Part of the submission process includes Pre-Submission (Pre-Sub) meetings with the FDA. These can be very valuable, but only if the feedback is taken seriously. We treated Pre-Sub input as a contract, not a suggestion.
Every item raised during those discussions was addressed explicitly, point by point, in the final submission. If you’re not prepared to incorporate or directly respond to Pre-Sub feedback, it’s fair to ask why you’re having the meeting at all. Alignment early pays off later.
Building Clinical Trust Through Regulatory Excellence
For PathPresenter, FDA 510(k) clearance is not just a regulatory checkbox. It reflects our commitment to building clinical software that pathologists can trust in real-world diagnostic workflows.
We’re proud of the clearance, and equally proud of the disciplined, transparent approach that got us there. As digital pathology continues to evolve, we remain committed to quality, safety, and innovation, grounded in a regulatory process that exists for a reason.
If you’d like to learn more about the PathPresenter Clinical Viewer or see it in action, we invite you to request a demo or reach out to our team.
More Posts
- PathPresenter Invites Pathologists to Expect More from their IMS with Image Biorepository Management Module at USCAP 2026
- Pathology Education: The Digital Future Is Here
- Why a Vendor-Agnostic IMS Is Key to a Future-Proof AI Strategy
- The ROI of Remote Consultations
- Digital Pathology Basics: What is Digital Pathology